ÖNEMLİ: Aşağıda okuyacağınız makalede bazı epilepsi türlerinde kullanılan ilaç isimleri yeralmaktadır. Bu ilaçlar doktor tavsiyesi olmadan kesinlikle kullanılmamalıdır. Yanlış kullanılan ilaçlar telafisi mümkün olmayan hasarlara sebep olabilir.
***
Epilepsi, beynin belirli bir süreliğine fonksiyonel bozukluğudur. Epileptik nöbetler ile karakterizedir. Nöbetler beyindeki sinir hücrelerinin (nöronlar) kısa bir süreliğine, aniden kontrolsüz olarak uyarılması ile ortaya çıkar ve şiddeti hastadan hastaya değişir. Nöbetler bazı hastalarda tek tek kaslarda yalnızca hafif bir seğirme veya karıncalanma şekline olurken bazı hastalarda orta şiddette sarsıntılar veya kısa süreli bilinç kaybı ile birlikte tüm vücutta kontrolsüz sarsıntılar şeklinde olur.
Epilepsinin nedenleri ve risk faktörleri
Epilepsinin çoğu vakada belirgin bir nedeni yoktur ve gelişimi karmaşık ve çeşitlidir. Bazı durumlarda, hastanın açık seçik epilepsi nöbeti geçirdiği bilinmesine ve modern muayene yöntemleri ile tespit edilmesine rağmen nöbetlerin spesifik bir nedeni tespit edilemez. Bu tür vakalara için kriptojenik epilepsi (açıklanamayan epilepsi) denir.
Bazı durumlarda nöbet geçiren hastanın beyninde metabolik bir bozukluk veya patolojik bir değişiklikliğe rastlanmaz. Böyle durumlarda hastanın neden epileptik nöbet geçirdiği açıklanamaz. Bu tür vakalara idiyopatik epilepsi denir. Ancak son zamanlarda, idiyopatik epilepsi terimi yerine genetik epilepsi terimi kullanılmaktadır. Bunun sebebi hastaların çoğunda, nörotransmitterlerin bağlanma bölgeleri olan reseptörlerde genetik değişikliklerin tespit edilmiş olmasıdır. Uzmanlara göre bu tür genetik değişiklikler epilepsi gelişimine katkıda bulunabilir fakat genellikle kalıtsal değildir. Ebeveynler genellikle nöbetlere karşı duyarlılıklarını çocuklarına aktarırlar ancak nöbetler çocukta uykusuzluk veya hormonal değişiklikler gibi dış etkenler ile tetiklenir.
Bunların dışında birçok hastanın beyninde oluşan yapısal değişiklikler veya çeşitli hastalıklar epileptik nöbetlere neden olabilmektedir. Bu durumdaki vakalara semptomatik epilepsi veya yapısal/metabolik epilepsi denir. Konjenital malformasyonlar (doğum öncesi dönemde oluşan beyin hasarları), hamilelik sırasında oksijen eksikliği, beyin kanaması veya vasküler malformasyonlar, bir kaza sonucu oluşan kafa travmaları, beyin tümörleri, felç, beyin iltihabı, menenjit, diyabet, tiroid gibi bazı metabolik hastalıklar, zehirlenme, alkol veya uyuşturucu kullanımı sonucu oluşan epilepsiler semptomatik epilepsiye örnek teşkil eder.
Sınıflandırma
International League Against Epilepsy (ILAE) (Uluslararası Epilepsiye Karşı mücadele Ligi), en son 1989’da onaylanmış olan sınıflandırmayı yeni bilimsel veriler ışığında 2017 yılında güncelledi. Buna göre hastalığın sınıflandırılması ve hastalıkta kullanılan terminoloji kısmen değişti.
International League Against Epilepsy (ILAE) nöbet tipine göre epilepsiyi dört temel sınıfa ayırdı:
- Genelleştirilmiş epilepsi
- Fokal epilepsi
- Genelleştirilmiş ve fokal epilepsi
- Genelleştirilmiş veya fokal epilepsi olup olmadığı belli olmayan epilepsi

Epilepsi türü nasıl teşhis edilir?
Epilepsi farklı semptom ve kalıpları olan kompleks bir hastalıktır. Bu nedenle epilepsinin birçok farklı türü vardır. Epilepsinin tipini sınıflandırmak ve hangi türe sahip olduğunu bilmek doğru tedaviyi bulmada önemlidir. Her epilepsi türünde değişik nöbet tipleri vardır ve bunlar beyinde normal elektriksel dalgalanmaları geçici olarak bozan/engelleyen gök gürültülü fırtınalar gibidirler.
International League Against Epilepsy (ILAE), yeni nöbet sınıflandırmasını üç temel özelliğe göre yapıtı.
- Beyinde nöbetlerin başladığı yer
- Nöbet sırasındaki farkındalık/bilinç düzeyi
- Nöbetlerin ve diğer semptomların özellikleri
Epilepside ortaya çıkan semptomları değerlendirmek ve buna göre hastalığı doğru bir şekilde sınıflandırmak oldukça zor ve uzmanlık gerektiren bir konudur. Bu yüzden hastanın tedavisinin uzman bir hekim tarafından yürütülmesi gerekmektedir.
Doktorlar, elektroensefalografi (EEG), nörogörüntüleme, genetik ve metabolik testler yaparak yüksek düzeyde teşhis koyabilirler. Doktorlar ayrıca hasta ve yakınlarına bazı sorular sorarak, eğer varsa nöbetler esnasında çekilen bir videoya bakarak en doğru tanıyı koymaya çalışır.
1- Nöbetleri beyinde başladığı yere göre tanımlama
Epilepsi tedavisinde kullanılacak ilaçlar, cerrahi müdahaleler ve diğer seçenekler nöbet çeşidinin tespiti için önemlidir. Bu nedenle sınıflandırmada ilk adım nöbetlerin beynin hangi bölgesinde başladığını tespit etmektir. Bunun için hastanın durumunun aşağıdaki seçeneklerden hangisine uyduğu bulunur.
- Fokal/odak nöbetler: Eski sınıflandırmaya göre kısmi nöbet olarak bilinen bu nöbetler beynin bir tarafında bulunan hücreler veya hücre ağlarında başlar.
- Genelleştirilmiş nöbetler: Eski sınıflandırmaya göre birincil jeneralize nöbetler olarak adlandırılan bu nöbetler, beynin her iki tarafındaki ağlarada başlar. Nöbetler genelleştirilmiş motor nöbet veya jeneralize motor olmayan nöbet şeklinde olabilir.
- Bilinmeyen nöbetler: Bir nöbetin başlangıç yeri bilinmiyorsa, nöbet bilinmeyen başlangıçlı kategorisine girer. (İlerleyen zamanda nöbetlerin başladığı yer netleştikçe nöbet tipi değiştirilebilir)
- Focal ve bilateral nöbetler (iki odaklı nöbet): Beynin bir tarafında veya bir kısmında başlayan ve daha sonra her iki tarafa yayılan nöbetlerdir. İkincil jeneralize nöbet olarak da bilinir.
2- Nöbet sırasında farkındalık/bilinç düzeyine göre tanımlama
Bilinç, kişinin nöbet sırasında vücut güvenliğini etkileyen ana faktörlerden biri olduğu için pratik öneme sahiptir. Değerlendirmesi daha kolay olduğu için bilinç yerine farkındalık kullanılır.
- Fokal farkındalık: Eğer kişi nöbet sırasında konuşamıyor veya tepki veremese bile, farkındalık bozulmadan kalıyorsa, bu nöbete fokal farkında nöbet
- Fokal bozulmuş farkındalık: Nöbet sırasında farkındalık/bilinç bozulmuş veya kişi kendisine ne olduğu hakkında belirsiz bir fikre sahipse bu tür nöbetlere fokal bozulmuş farkındalık (Bu terimdir, 2017 yılından önce karmaşık kısmi nöbet terimi kullanılıyordu.
- Bilinmeyen farkındalık: Kişi yalnız yaşıyorsa veya sadece geceleri nöbet geçiriyorsa kişinin nöbet sırasında bilincinin yerinde olup olmadığını bilmek mümkün değildir. Bu durumda farkındalık terimi kullanılmaz onun yerine bilinmeyen farkındalık terimi kullanılır.
- Genelleştirilmiş nöbetler: Nöbetlerin bilinci veya farkındalığı bir şekilde etkilediği varsayılır. Bu nedenle genelleştirilmiş nöbetlerde farkındalığı tarif etmek için özel terimlere gerek yoktur.
3- Fokal/Kısmı nöbetlerde motor ve diğer semptomları tanımlama
Nöbet sırasında birçok değişik semptom ortaya çıkabilir. Bu da nöbetlerin temel sistemde özelliklerine göre alt gruplara ayrılmasına sebep olur.
- Fokal motor nöbet: Nöbet sırasında değişik dramatik hareketler meydana gelir. Örneğin vücudun bir parçasının seğirmesi, sarsılması veya sertleşmesi veya dudak yalama, elleri ovuşturma, yürüme veya koşma gibi otomatik hareketler ortaya çıkabilir (otomatizmalar)
- Fokal motor-olmayan nöbet: Bu tür nöbetlerde nöbet öncesi semptomlar vardır, Örneğin, duygu, düşünce veya deneyimlerde değişiklikler gibi…
Auralar, nöbet başlangıcının hemen öncesinde hissedilen semptomlardır. (Aura terimi yeni sınıflandırmada yer almıyor ama yine de bu terim kullanılmaya devam ediyor zira bu erken semptomların bir nöbet başlangıcı olabileceğini bilmek önemlidir.)
***
Çocukluk çağı epileptik sendromları
Syndrom terimi, genellikle farklı semptomların bir kombinasyonu anlamına gelir. Çocukluk çağı epilepsi ise belirli bir yaşta başlayan ve farklı nöbet kombinasyonları gösteren bir tür çocukluk sendromu olarak kabul edilir.
Eğer bir hastaya genetik analizler, EEG sonuçları ve hastalığın seyrindeki tipik özellikler değerlendirilerek epilepsi tanısı konmuş ise bir uzman hekim eşliğinde uygun bir tedaviye geçilir.
Epileptik ensefalopatiler (EE): Epileptik ensefalopatiler, genellikle hayatın ilk bir yıllarında başlayan ve ilâçlı tedaviye dirençli epileptik sendromları tanımlamak için kullanılır. Epileptik ensefalopati ayrıca beyindeki fonksiyonel bozuklukları da tetiklediği durumları ifade eder. Dolayısıyla epileptik ensefalopati sadece epileptik nöbetlere yol açmazlar aynı zamanda gelişimsel ve dil bozukluklarına da yol açabilir. Epileptik ensefalopatilerin mutlaka olumsuz bir seyir izler diye bir kural yok, bazen süreç iyi doğru da evrilebilir.
Yenidoğanlarda epilepsi sendromları
Erken çocukluk çağı epileptik ensefalopatisidir. Çok nadir görülen çok ciddi bir epilepsi sendromudur.
Ohtahara sendromu, kontrol edilmesi zor nöbetler ve gelişimsel gecikmeler ile karakterizedir. Epileptik nöbetler genellikle yaşamın ilk üç ayında (çoğunlukla doğumdan sonraki ilk 10 gün içinde) ortaya çıkar.
Nöbetler, sık tonik spazmlar (kasların çok gergin kaldığı uzun süreli spazmlar) veya fokal-motor (beyinde bir bölgede sınırlı) veya nadiren miyoklonik nöbetler (istemsiz kas hareketi) şeklindedir.
MRT (Manyetik rezonans görüntüleme) tekniği ile yapılan çekimlerde etkilenenlerin bazılarında beyin malformasyonları bulunmakla birlikte büyük bir çoğunluğunda hiçbir değişikliğin olmadığı görüldü. Bu sendromun seyri kötüdür. Çoğunlukla ağır engelli olan çocukların yaklaşık her ikisinden biri hayatlarının ilk yılında ölmektedir. Son çalışmalar, Ohtahara sendromunun genellikle genetik nedenlerden kaynaklandığını göstermektedir.
Ohtahara Sendromunun genetik nedenleri
Ohtahara sendromu vakalarının çoğu, beyin malformasyonundan veya belirli gen mutasyonlarından kaynaklanır. Metabolik nedenler daha az olasıdır. Bazı durumlarda, net bir neden bulunamaz.

Beyin malformasyonlar yaygın olarak beynin her iki tarafında görülmekle beraber beynin yalnızca bir alanında veya bir tarafında olabilir.
Gen mutasyonları, bir tek gende veya birden fazla gende olabilir. Ohtahara sendromuyla ilişkilendirilen bazı genler şunları içerir: ARX, CDKL5, SLC25A22, STXBP1, SPTAN1, KCNQ2, ARHGEF9, PCDH19, PNKP, SCN2A, PLCB1, SCN8A, ST3GAL3, TBC1D24, BRAT1
Tedavi: Nöbetleri kontrol etmek için nöbet önleyici ilaçlar kullanılır ancak bu ilaçların çok etkili olduğu söylenemez. Kortikosteroidler (prednizolon veya ACTH) bazı hastalarda faydalıdır. Bazen ketojenik diyet (yüksek yağ, düşük karbonhidrat) uygundur.
Fokal beyin lezyonu olduğu durumlarda (beynin bir bölgesinin hasar görmesi) cerrahi müdahale faydalı olabilir
West sendromu, genellikle yapısal beyin hasarının (ensefalopatinin) neden olduğu, nadir görülen bir erken çocukluk dönemi epilepsisi dir. Semptomlar, vakaların %50-70 inde yaşamın 3. ve 7. ayı arasında başlar. Nadir de olsa doğumdan hemen sonraki günlerde veya 5 yaşına doğru da başlayabilir.
Hastalığın prevalansı 1- 6 / 100.000 olarak tahmin edilmektedir. Bazı kaynaklarda daha fazla olduğu rapor edilmiştir.
Blitz-Nick-Salama-Kramps (BNS) olarak da bilinen bu epilepsi türünde nöbetler, şimşek gibi kısa kasılmalar veya tonik spazmlar (kasların çok gergin kaldığı uzun süreli spazmlar), başın ve vücudun üst kısmının öne doğru bükülerek kolların yukarıya doğru kaldırılması ile karakterizedir. Bu kramplar 0,2 ila 2 saniye gibi sürer. Ancak diğer nöbet türleri de mümkündür.
West Sendromu vakalarının yaklaşık %75 i beyinde meydana gelen malformasyonlar veya metabolik bozukluklardan kaynaklanırken %25 nin neden bilinmiyor. Erkekler hastalıktan kızlara göre daha sık etkilenir. Ayrıca Down sendromlu çocukların West sendromu geliştirme riski daha yüksektir.
Genetik yatkınlık: Yapılan araştırmalar beyin gelişimi ve işlevinde rol oynayan ARX ve CDKL5 genlerinin mutasyonlu formlarının erken çocukluk çağı nöbetlerine neden olduğunu gösterilmiştir. Her iki genin X kromozomu üzerinde bulunması nedeni ile hastalık X bağlantılı West sendromu olarak da bilinir.
- ARX genindeki mutasyon: ARX genindeki mutasyondan kaynaklanan West sendromu, X’e bağlı resesif kalıtımdır yani genin iki kopyasından birinin mutasyonlu olması sendromu tetiklemek için yeterlidir. Erkekler için durum daha ciddidir zira X kromozomu erkeklerde bir tanedir dolayısı erkeklerde bu genin sağlıklı bir kopyası bulunmaz dolayısı ile kadınlara göre semptomlar daha ağırdır.
- CDKL5 genindeki mutasyon: CDKL5 genindeki mutasyondan kaynaklanan West sendromu, X’e bağlı dominant kalıtımdır yani sendromu tetiklenmesi için genin iki kopyasının da mutasyonlu olması gerekli. CDKL5 geninde meydana gelen mutasyon hem erkeklerde hemde kadınlarda sendromun eşit şiddette görülmesini sebep olur.
Bu genlerin dışında daha başka genler de rapor edilmiştir.
Tedavi: West sendromunun tedavisi için tek tip bir terapi konsepti yoktur.
İlaç tedavisi şunları içerir
- Adrenokortikotropik hormon (ACTH) veya glukokortikoidler
- Antikonvülsanlar (sultiam, vigabatrin, levetirasetam, valproat, topiramat)
- Benzodiazepinler (Klonazepam, Nitrazepam)
- Piridoksal fosfat (B6 vitamini)
İlaçlı tedaviye yanıt kişiden kişiye değişir. Hastalık ilaçlı tedaviye dirençlidir.
Operatif tedavi
Beyin hasarı durumunda cerrahi müdahale düşünülebilir. Tedavi başlangıçta başarılı olmasına rağmen, 6 ay sonra vakaların %30 unda hastalık yeniden başladığı rapor edilmiştir.Çocuklar 5 yaşına geldiğinde nöbetler genellikle durur ancak daha sonra nüks ettiği rapor edilmiştir. Motor, duyusal eksiklikler yeni yürümeye başlayan çocukların %75 inde görülür ve epilepsi vakaların %50-60 ında tedaviye dirençlidir.
- Dravet-Syndrom: Bu konuda ayrıntılı makaleye buradan ulaşabilirsiniz
Küçük çocuklardaki epilepsi sendromları
- Lennox-Gastaut sendromu (LGS)
Lennox-Gastaut sendromu (LGS), nöbetler ilk kez 2 ile 7 yaşları arasında ortaya çıkar ve çok farklı biçimleri vardır. Hastalığın yaklaşık yüzde 70-80 nin tanımlanabilir bir nedeni vardır. Hastalık semptomatik lennox-gastaut sendromu olarak da adlandırılır. Sebepler arasında beyin korteksinin anormal gelişimi (kortikal displazi), konjenital enfeksiyonlar, inme, travma, doğumdan önce meydana gelen düşük oksijen (perinatal hipoksi), ensefalit gibi merkezi sinir sistemi enfeksiyonları yer alır. Menenjit ve tüberoskleroz veya nadir bir genetik bozukluklar da sebepler arasında yer almaktadır.
Lennox-Gastaut sendromlu bireylerin yaklaşık yüzde 17-30 unda daha önce West sendromu öyküsü vardır. Hastaların yaklaşık yüzde 20-30 unda neden bilinmiyor. Bu grup Kriptojenik Lennox-Gastaut sendromu olarak da sınıflandırılabilir. Bu grupta olan kişilerde önceleri nöbet aktivitesi, nörolojik problemler veya bilişsel bozukluk öyküsü yoktur. Kriptojenik vakalarlarda semptomlar genellikle daha geç başlar.
Lennox-Gastaut sendromunda nöbetler sıklıkla uyku sırasında ortaya çıkar ve Lennox-Gastaut sendromunun en karakteristik özelliği tonik kramplar (%17-92), atonik kramplar (Kasların ani bir şekilde gevşemesi sonucu düşme ve kısa süre sonra tekrar kalkma) (%26-56) ve atypical absences nöbetlerdir (%20-65). (beynin her iki tarafında başlayan tipik olmayan genel başlangıçlı nöbetler)
Tedavi: Tedavi zordur çünkü geleneksel antikonvülsanlar genellikle başarısızdır. Tedavisinde, valproat, lamotrijin, felbamat veya topiramat gibi antiepileptik ilaçları uygulanır. Genellikle nöbetleri kontrol edecek tek bir antiepileptik ilaç yoktur. Çocuklar başlangıçta iyileşebilir ancak bir çoğu daha sonra ilaca tolerans gösterir veya kontrol edilemeyen nöbetler geliştirir.
Hasta deneyimli bir uzman tarafından muayene edilerek en uygun ilaç ve doz belirlenmelidir ve etkili ilaç tespit edildikten sonra ilaç düzenli olarak alınmalıdır.
Ebeveynler ve çocuğun düzenli uyumasına ve uyku ritmine dikkat etmeli ayrıca nöbetleri tetikleyebilecek parlak yanıp sönen ışıklardan ve karmaşık motiflerden kaçınmalıdır. Çocuğun düşmesi durumunda kendini çok fazla yaralamaması için kask takmasında fayda vardır.
Ketojenik diyet: Özellikle 1930’dan beri yüksek yağlı ve düşük karbonhidratlı ketojenik diyet bilinen ek bir tedavi şeklidir. 2000 li yıllardan itibaren özellikle epilepsisi ilaçlarına yanıt vermeyen çocuklarda bu terapi giderek daha sık kullanılmaya başladı.
Vagus sinir stimülasyonu: Bazı hastalarda vagus sinir stimülasyonu ile tedavi denenebilir. Bunun için köprücük kemiği üzerindeki derinin altına cerrahi bir müdahale ile küçük bir cihaz yerleştirilir ve elektriksel uyarılar gönderen elektrotlar ile vagus sinirleri uyarılır.
Hastalığın genel popülasyonda yıllık insidansının 1:1.000.000, prevalansının ise 15:100.000 olduğu tahmin edilmektedir. Etkilenen çocuklar genellikle fiziksel ve zihinsel gelişim gecikmeleri gösterir.
Doose sendrom, Miyoklonik astatik epilepsi olarak da bilinir. 18 ay ile 5 yaş arasında başlar. Miyoklonik-astatik ve Atonik-astatik nöbetler bu sendromun karakteristiğidir. Çoğu çocuk ateşsiz olarak jeneralize tonik-klonik nöbetler (kaskatı kesilip ve yere düşme ve vücut kaslarında kasılıp gevşeme) geçirir. Ateşe bağlı nöbetler %11 ila 28 oranındadır.
Teşhis yöntemleri: Nöbet türü, nörolojik muayene ve elektroensefalogram (EEG) bulgularına dayanır. Ayırıcı genetik ve metabolik testler yapılmalıdır. Vakaların bir çoğunda neden tam olarak bilinmemekle beraber poligenik bir kalıtımdan şüphelenilmektedir. En yaygın monogenik nedenler arasında SLC6A1 (3p25.3), CHD2 (15q26.1), AP2M1 (10q23.2) bulunur. Bildirilen diğer nedensel değişkenler arasında SLC2A1 (1p34.2), SCN1A (2q24.3), SYNGAP1 (6p21.32), KCNA2 ve NEXMIF (Xq13.3) genleri de bulunmaktadır.
Doose sendromunun prognozu (hastalığın seyri) karışıktır. Araştırmalar Doose sendromlu çocukların yaklaşık yüzde

20 sinde gelişimsel gecikme olduğunu, eğer çocuklar nöbet geçirmiyorsa, zihinsel ve psikomotor gelişimlerinin olumlu olduğunu gösteriyor.
Tedavi: Bazı çocuklarda nöbetler başarılı bir şekilde tedavi edilebiliyor ve bu çocuklar normal bir gelişim gösteriyor. Bazı çocuklarda ise nöbetleri kontrol etmek zordur. Gelişim bozukluğu gösteren bu çocuklar sıklıkla nöbet önleyici ikili veya üçlü kombine ilaçlara ihtiyaç duyarlar.
Sodyum valproat, sodyum valproat ile lamotrijin kombinasyonu sinerjistik etki gösteriyor. Özellikle miyoklonik nöbetlerde levetirasetam ve etosüksimid etkili olduğu, alternatifler olarak da zonisamid, topiramat, klobazam ve klonazepam etkili olduğu belirtiliyor.
Nöbet önleyici ilaçların başarısız olduğu durumlarda ise ketojenik diyet öneriliyor. Ketojenik diyetin, miyoklonik-astatik epilepside en etkili sonuç verdiği ve bu tedavi ile hastaların %50-90 ında %50 oranında nöbetlerin azaldığı rapor edilmiştir.
Prognoz (hastalığın seyri): Sonuçlar değişkendir. Hastaların %60 ında normal bilişsel gelişme veya hafif bir bilişsel gecikme görülür. Üç yıl ve üzeri devam eden nöbetlerde bilişsel gelişim ve davranışlarda olumsuzluklar görülür.
Yaşa bağlı olarak gelişen bir epileptik ensefalopati çeşididir. Nöbetler, 2-4 yaşları arasında başlar ve genellikle uykuda ve vücudun bir tarafında ortaya çıkar. Nöbetler tonik-klonik veya klonik türdedir. Nöbetler 5-6 yaş civarında hem daha sık hem daha şiddetli hem de tedaviye dirençli hale gelir. Bu dönemde gelişimde belirgin bozulmalar olur. (Örneğin. dil, sosyal etkileşim, genel zeka, motor beceriler ve davranışlarda gerileme gibi). Ergenlik öncesi nöbetlerde ve EEG deki anormalliklerde spontan bir düzelme olsa da çoğu hasta ciddi şekilde geri kalır.
Teşhis: Vakaların yaklaşık % 50 sinde talamusu etkileyen vasküler hasarlar veya kortikal gelişim malformasyonları gibi erken gelişimsel bozukluklar tanımlanmıştır.
Genetik faktörler: Yakın zamanda CSWS ile ilişkilendirilmiş GRIN2A geninde mutasyonlar bulunmuştur. CSWS nin otozomal dominant kalıtım tarzında çocuğa aktarıldığı düşünülüyor. Bu nedenle CSWS sendromlu çocuğu olan ebeveynlerin en az birinde GRIN2A geninin mutasyonlu olduğundan şüpheleniliyor. Bu nedenle ailelere genetik danışmanlık önerilir.
Tedavi: Tedavinin temel amacı nöbetleri kontrol etmektir. EEG deki anormalliklerin uzun vadede iyileşip iyileşmeyeceğini tahmin etmek zordur. Diazepam veya klobazam gibi yüksek dozlarda gece alınan benzodiazepinler, epileptik aktiviteyi azaltır. En sık kullanılan antiepileptik ilaçlar valproat, levetiracetam, lamotrigine ve ethosuximid tır. Kortikosteroidler faydalıdır, ancak uzun vadede yan etkileri vardır. Bazı özel vakalarda cerrahi müdahale etkili bir tedavi yöntemi olabilmektedir.
Hastalığın Prevalansı bilinmiyor. Bazı çalışmalara göre epilepsili çocukların % 0.5-1.5’ni etkileyen nadir bir sendromdur. Erkekler hastalıktan kızlara göre daha sık etkileniyorlar (3:2).
Prognoz (hastalığın seyri): Ergenlikle birlikte nöbetler ve EEG deki anormallikler normale dönme eğiliminde olsa da çoğu vakada bilişsel bozulma devam ettiği için gelişimin prognozu kötüdür.
Landau-Kleffner sendromu, genellikle üç ila on yaşları arasında ortaya çıkar ve sadece çocukları ve ergenleri etkiler. Etkilenen çocukların yaklaşık %70 i çeşitli türde epileptik nöbet geçirirler ve bunların çoğunluğu uykuda gerçekleşir.
Landau-Kleffner sendromu, nadir görülen kombine fokal generalize epilepsidir (Nedeni bilinmeyen genelleştirilmiş nöbetler ile karakterize bir epilepsi türü) ve konuşma merkezinde meydana gelen bir hasar sonucu oluşan dil

bozuklukları hastalığın tanımlayıcı özelliğidir (edinilmiş afazi). Konuşma zorluklarının yanı sıra okuma veya yazma gibi becerilerde de kısmen ya da tamamen zorluklar görülür. Çoğu zaman, dil becerileri günler veya haftalar içinde aniden kaybolur. Her dört çocuktan sadece biri dil becerilerini birkaç ay içinde yavaş yavaş geri kazanır.
Hastalığın ilk belirtileri, duyulanı anlamada yaşanan güçlüklerdir. Bazı çocuklar tamamen konusamazken bazı çocuklar konuşma akıcılığını kaybederler.
Dilsel gerilemeye ek olarak, dikkat eksikliği, hiperaktivite, dikkat dağınıklığı gibi davranış sorunları da yaygındır. Hastaların üçte ikisinde nöbetler görülür ve bu nöbetlerin kontrol edilmesi genellikle kolaydır. Nöbetler 15 veya 16 yaşına kadar yani ergenlik döneminde kaybolabilir ancak etkilenenlerin üçte ikisinde yetişkinlikte bile az çok ciddi dil bozuklukları görülür.
Hastaların yaklaşık üçte birinde cidde bir nöbetler olmasada kısmi motor nöbetler sıklıkla gözlenebilir.
Teşhis yöntemleri: Landau-Kleffner sendromunu güvenilir bir şekilde teşhis etmek için çeşitli nöropsikolojik yöntemler kullanılır. Örneğin, bir EEG çekimi yapılır. Hasta uyanıkken yapılan EEG çekimi genellikle herhangi bir bulgu vermez ancak uykuda yapılan EEG çekimi bulgular konusunda yardımcı olur. Ayrıca hastanın klinik görünümü örneğin, edinilmiş afazi özellikleri de (konuşma ve anlama zorluğu) tanıda yardımcı olan özelliklerdir.
GRIN2A mutasyonu olan ailelerde otozomal dominant kalıtımdan şüphelenilmelidir.
Tedavi: Nöbetleri önlemede kullanılan birçok antiepileptik ilaç genellikle etkilidir. Bu nedenle nöbet kontrolü sorun değildir. Nöbetler, antikonvülzanlar (valproat, karbamazepin, sultiam) veya glukokortikoidler kullanılarak ilaçla tedavi edilir. Ayrıca Kortikosteroidler dil becerilerini geliştirmek ve stabilize etmekle de ilişkilidir. Konuşma ile ilgili sonuçlar hastadan hastaya değişkendir. Dil becerileri ergenlikle gelişebilir ancak hastalık öncesi aşamaya ulaşamaz. Hastalara işitme testleri ve takip muayeneleri önerilir. Konuşmayı anlama, geliştirme ve sürdürme terapisi gereklidir. Bazı hastalarda cerrahi bir teknik olan subpial transeksiyonu başarıyla kullanılmıştır.
Prognoz (hastalığın seyri): Nöbetlerin kontrol edilebilmesi ve ergenlikle birlikte kaybolması hem hasta hem de hasta yakını için olumlu bir durumdur. Hastalığın seyrindeki farklılıklar nedeniyle genel gidişat ile ilgili iddialı bir şey söylemek mümkün değildir. Nöbetler durduktan sonra semptomlarda iyileşmeler prensip olarak mümkündür ancak tam remisyon (belirti ve bulguların ortadan kalkması) çok nadirdir.
Panayiotopoulos sendromu, çocukluk çağında nispeten yaygın görülen bir epilepsi türüdür. Hastalık öyküsü doğum öncesi ve doğum sonrası normaldir. Doğum sonrası yapılan muayeneler kafa büyüklüğü ve diğer testler genellikle normaldir.
Hastalığın erken varyantı 2 ila 8 yaşları arasında başlarken geç varyantı 3 ila 16 yaşları arasında başlar. Etkilenenlerin üçte birinde nöbet süresi değişkendir. Nöbet süre kimi zaman dakikalar, kimi zaman saatler sürebilir. Gece uyku esnasında gelen nöbetlerde yapılan EEG çekimlerinde nöral aktiviteler genellikle yüksek tepelidir. Çoğu hastada nöbetler nadir görülür ve bu hastaların %25 inde tek bir çeşit nöbet görülür. Az bir hastada ise nöbetler sık görülür bunlar otonom status epileptikus şeklinde nöbetler olabilir. (uzun veya seri aralıklarla olan kısa nöbetler) Genellikle 11-13 yaşlarda nöbetlerde düzelme görülür. Hastaların %5-17 sinde ise ateşli havale öyküsü vardır.

Yaklaşık her dört vakanın birinde aile öyküsü vardır (epilepsi veya başka nöbetler türleri). Panayiotopoulos sendromu her iki cinsiyet de eşit oranda görülür. Gelişim ve biliş normaldir. Bununla birlikte aktif nöbet dönemlerinde konuşmada sorunlarının olduğu rapor edilmiştir.
Panayiotopoulos sendromu, beynin arkasında yer alan loblardan biri olan oksipital lobdan kaynaklanan fokal epilepsi lerden biridir. Nöbetler daha çok otonom sinir sistemini etkileyen fokal nöbetlerdir (beynin sınırlı bölgesi). Etkilenen çocukların dörtte üçü mide bulantısı ve kusmadan şikayet ederler ve solgun görünürler. Göz bebekleri genellikle ciddi şekilde daralır veya genişler.
Tedavi: Panayiotopoulos sendromlu çocuklarda nöbetler genellikle seyrek görülür bu nedenle nöbet önleyici ilaçlara ihtiyaç duyulmayabilir. Nöbetlerin daha sık olduğu durumlarda nöbet önleyici ilaçlar için doktor ile görüşülmelidir
Çocukların uzun nöbet geçirmesi durumunda, acil tıbbi tedaviye ihtiyaç olabilir. Diazepam rektal jel veya dilin altında verilen benzodiazepin türü preparatlar tedavide yardımcı olabilir.
Ebeveynleri, acil durumlar hakkında bilgi edinmek için çocuğu tedavi eden nörolog veya sağlık uzmanıyla konuşmalıdır.
Prognoz (hastalığın seyri): Panayiotopoulos sendromunun seyri odukça iyidir. Genellikle hastalık 14 yaşına kadar iyileşir.
Benzer konularda hazırlanmış diğer makaleler
Mehmet Saltuerk
++++++++++++++++++++++++++
Dipl. Biologe Mehmet Saltürk
The Institute for Genetics
of the University of Cologne
++++++++++++++++++++++++++
Kaynaklar
- ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology
- ILAE Official Report: A practical clinical definition of epilepsy
- Recent advances in the molecular genetics of epilepsy
- SYNGAP1 encephalopathy: A distinctive generalized developmental and epileptic encephalopathy
- De novo loss of function mutations in KIAA2022 are associated with epilepsy and neurodevelopmental delay in females
- Effect of Cannabidiol on Drop Seizures in the Lennox-Gastaut Syndrome.
- Infantile, Childhood, and Adolescent Epilepsies.
- Epilepsy with myoclonic-atonic seizures (Doose syndrome): Clarification of diagnosis and treatment options through a large retrospective multicenter cohort
- Efficacy and Safety of a Ketogenic Diet in Children and Adolescents with Refractory Epilepsy-A Review
- [Effectiveness of a ketogenic diet in children with refractory epilepsy: a systematic review].
- Ketogenic diet in the treatment of epilepsy in children under the age of 2 years: study protocol for a randomised controlled trial.
- Vagus nerve stimulation for drug-resistant epilepsy
- Vagus nerve stimulation for epilepsy.
- Clinical spectrum of SCN2A mutations expanding to Ohtahara syndrome
- https://www.mayoclinic.org/diseases-conditions/epilepsy/symptoms-causes/syc-20350093
- https://www.epilepsy.com/learn/about-epilepsy-basics/what-epilepsy
- https://epilepsysociety.org.uk/
- https://my.clevelandclinic.org/health/diseases/17636-epilepsy
- https://www.epilepsydiagnosis.org/
- https://www.ilae.org/guidelines/definition-and-classification/ilae-classification-of-the-epilepsies-2017
- Landau-Kleffner syndrome
- Neurocognitive and behavioural profile in Panayiotopoulos syndrome


























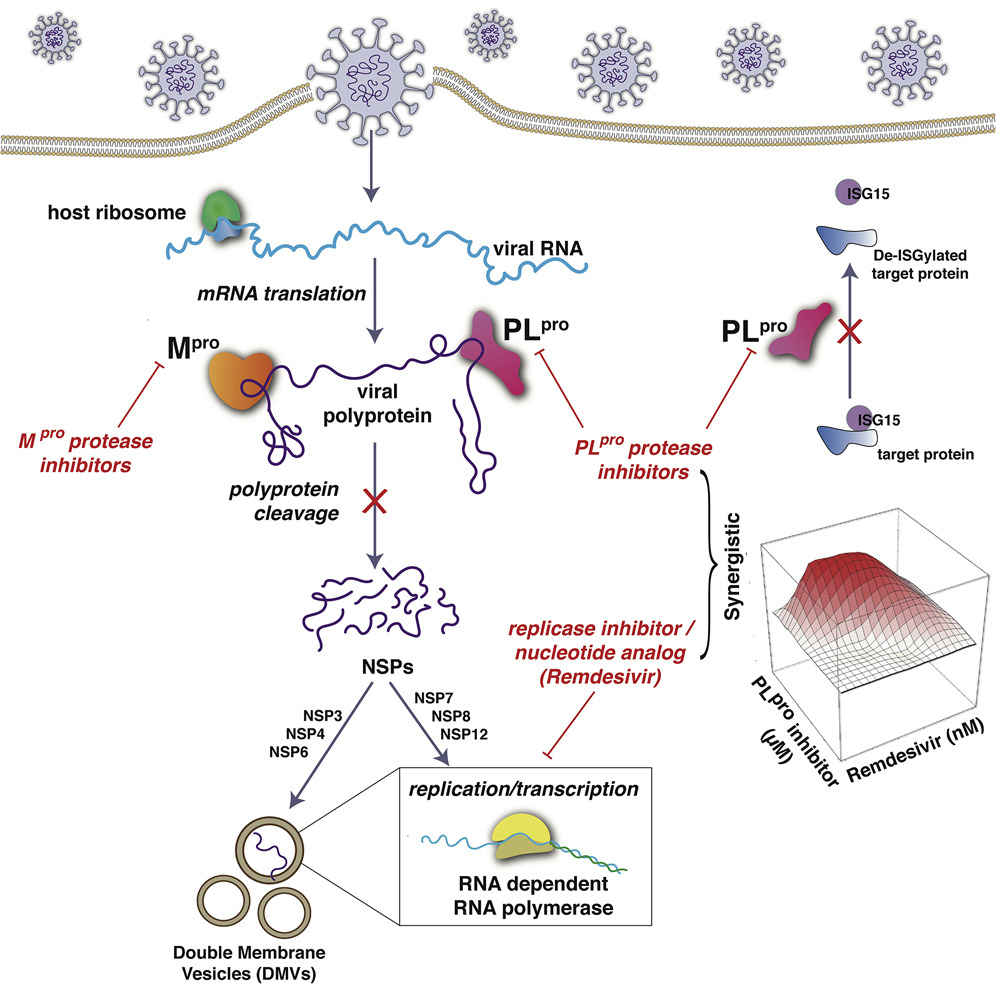
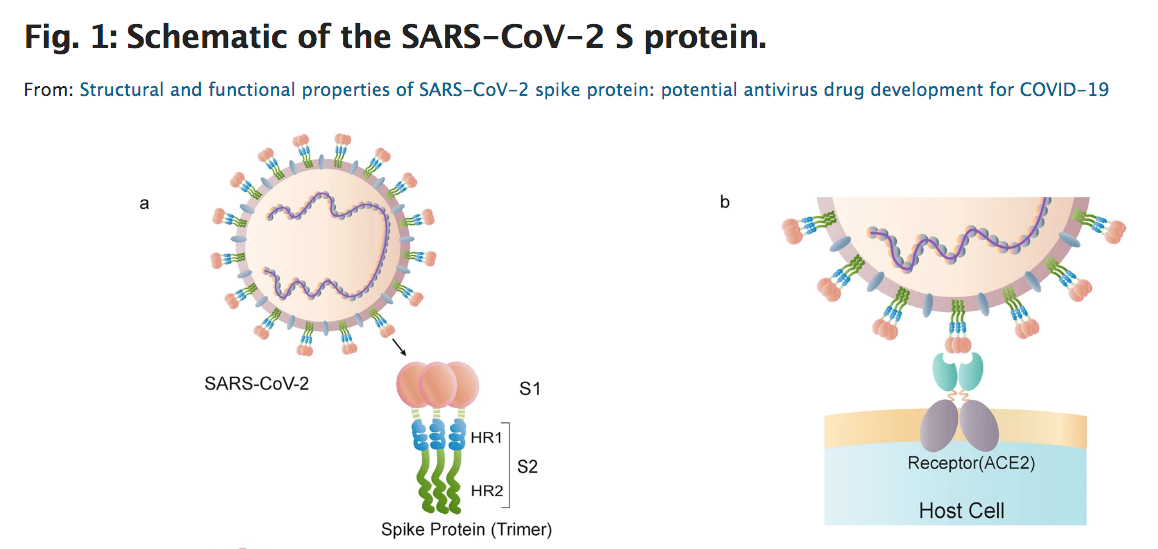 SP6 aptameri spike proteine bağlanarak enfeksiyonu önlüyor ama bunun nasıl olduğu hâla net olarak anlaşılmış değil. Zira SP6 aptameri, spike proteinin S1 kısmında bulunan ve hücreye kenetlendiği RBD (Rezeptorbindesdomäne) kısmına değil de S2 denen aşağı kısmına bağlanıyor. Oysa burası antikorların virüse bağlanarak virüsü etkisiz hale getirdiği kısım. Nasıl oluyorda SP6 aptameri bağlanma bölgesi RBD’ye değilde fonksiyonel bir bölge olan S2 bölgesine bağlanarak virüsün hücreye girmesini engelliyor. Bu garip fenomen hâla çözülebilmiş değil.
SP6 aptameri spike proteine bağlanarak enfeksiyonu önlüyor ama bunun nasıl olduğu hâla net olarak anlaşılmış değil. Zira SP6 aptameri, spike proteinin S1 kısmında bulunan ve hücreye kenetlendiği RBD (Rezeptorbindesdomäne) kısmına değil de S2 denen aşağı kısmına bağlanıyor. Oysa burası antikorların virüse bağlanarak virüsü etkisiz hale getirdiği kısım. Nasıl oluyorda SP6 aptameri bağlanma bölgesi RBD’ye değilde fonksiyonel bir bölge olan S2 bölgesine bağlanarak virüsün hücreye girmesini engelliyor. Bu garip fenomen hâla çözülebilmiş değil. Yaygın kullanılan bir astım spreyi şiddetli Covid-19 seyrini önlemede yardımcı olabilir.
Yaygın kullanılan bir astım spreyi şiddetli Covid-19 seyrini önlemede yardımcı olabilir.